
Pemphigus Vulgaris in a Black Patient: Early Recognition of Disease Saves Lives
María A. Rodríguez-Santiago*, Javier García-Marín, Alfredo Lamela-Domenech, María Vega-Martínez
Internal Medicine, School of Medicine, University of Puerto Rico, USA
Abstract
There is a well-known shortage of racial diversity in medical textbooks and literature contributing to race-based health care inequalities1. We present the case of a black puertorrican 58-year-old female who developed a painful non-pruritic blistering ulcer in the inner oral mucosa with associated erythema six months prior to the evaluation. She was misdiagnosed on multiple occasions leading to a rapid progression of the disease, and subsequently, her death. Lack of images in medical textbooks and scarce literature describing initial presentation per-skin-tone of Pemphigus Vulgaris (PV) in patients with dark skin color led to misdiagnosis, delay in treatment, and thus, this catastrophic outcome. This case report describes the appearance of PV in patients with dark skin tone and serves as an educational resource by providing images of a rare skin disease in people with dark skin. The purpose of this case report is to fill major gaps in medical literature, highlight the importance of timely recognizing PV in patients with dark skin, and to create awareness among physicians.
Introduction
Pemphigus vulgaris (PV) is a rare autoimmune disease (IgG and C3) that attacks transmembrane glycoprotein of the Cadherin family called Desmogleins 1 and 3. These proteins are an integral part of the epidermal desmosomes in the squamous stratified epithelium, working in the cell-to-cell adhesion of suprabasilar keratinocytes. The loss of cohesion between cells is called acantholysis, which clinically manifests as the formation of flaccid blisters2. This phenomenon can be seen histologically, giving the appearance of “row of tombstones”3.
Every year, PV incidence varies from 0.1-0.5 cases per 100,000 population with a higher prevalence in Europe and United States of America3. PV onset is usually between the fourth to sixth decade of life and is more common in Jewish or Mediterranean descendants4. Several retrospective studies referred that prevalence is equal in male and females, although other studies suggest 2:1 and, even, 3:1 female: male ratio5. Little is known about the prevalence or incidence of PV in dark skin tone or racial minorities.
PV has a high morbidity and mortality, and if left untreated disease will progress steadily with an average time of death of 14 months5. Therefore, early diagnosis is imperative. According to Risser et al., mortality for black persons was significantly higher for every pemphigoid bullous disease when compared to white people6. Interestingly, PV rarely presents in black ethnicity6. We believe that discrepancy between low prevalence of disease, but high mortality rate could be improved by teaching physicians how to competently diagnose PV in patients with dark skin tone.
There is a well-known shortage of racial diversity in medical textbooks and literature contributing to race-based health care inequalities1. The lack of images and evidence within literature concerning Pemphigus Vulgaris (PV) in patients with dark skin tone leads to a delay in diagnosis, thus having a negative impact on the patients’ management and overall prognosis.
Objective
We present the case of a 58-year-old black puertorrican female with a rapidly progressing PV who was misdiagnosed in multiple occasions, leading to a fatal result. This case aims to create awareness among Internists, Hospitalists and Primary care physicians (PCP), helping them recognize PV in dark skin patients, which is essential to improve outcomes.
Case Report
This is the case of a black 58-year-old female with medical history of tobacco and alcohol abuse who developed a “rash”, described as a painful non-pruritic blistering ulcer in the inner oral mucosa with associated erythema and brown-yellowish crust around the lips beginning six months prior to our evaluation. During that time, the patient visited her PCP and multiple Emergency Rooms receiving the diagnosis of Impetigo treated with topical Mupirocin. Rash progressed in a craniocaudal distribution involving the chest, abdomen and back. Subsequently, it worsened with dysphagia, odynophagia, poor oral intake, weight loss and general malaise. She denied recent antibiotic use, traveling, exposure to chemical agents, changes in diet or sick contacts at home. Physical examination showed large eroded and crusted plaques at different stages of healing over more than 30% of body surface (Figure 1a, b). Both, oral and conjunctival mucosa, had extensive ulcerative lesions over an erythematous base with crusting and serosanguineous fluid (Figure 2). Complete blood count (CBC) revealed leukocytosis with neutrophilic predominance and bandemia (WBC: 21.8 #/uL, Neutrophils: 79%, Bands: 18%). Arterial blood gases (ABGs) showed hypoxemia (pO2: 54.3 mmHg) for which she was placed in a nasal canula at 3L. Skin biopsy revealed acantholysis at the suprabasilar layer and Direct immunofluorescence (DIF) was positive for IgG deposition, both consistent with the diagnosis of PV. In view of extensive disease, she was started on high dose steroids pulses and intravenous immune globulin to complete five days each. Rituximab therapy was contraindicated due to ongoing bacteremia with multiple drug resistant organisms for which broad spectrum antibiotics were started. Skin lesions improved within days, showing re-epithelialization and post inflammatory dyspigmentation (Figure 3). Unfortunately, infection, malnutrition, immunosuppression and catabolic state, led the patient into septic shock requiring, vasopressors and mechanical ventilation. The patient died after twenty-one days at the Intensive care unit.
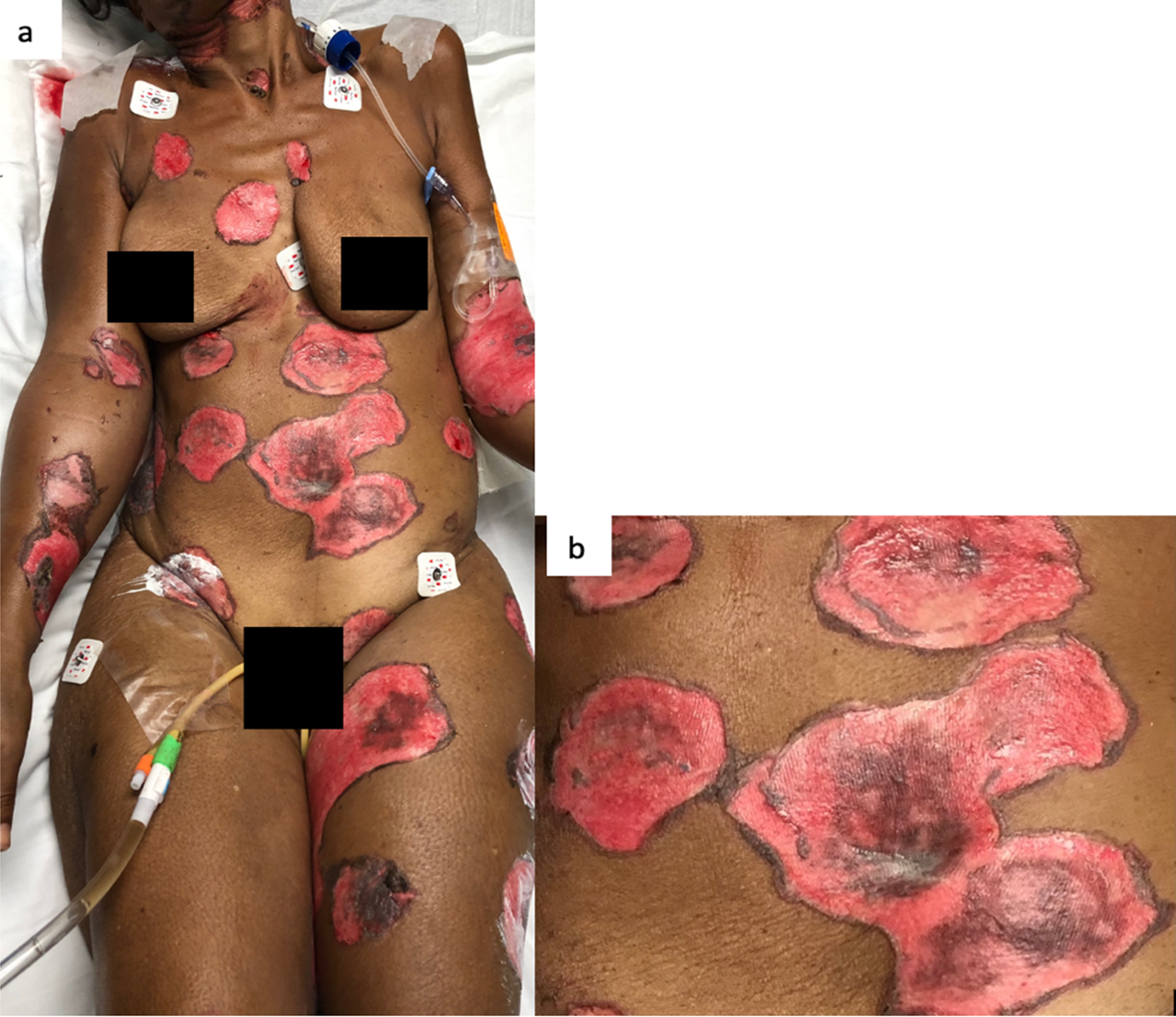
Figure 1: a) The skin involvement was over more than 30% of body surface. b) Large circular eroded and crusted plaques with hyperpigmented borders.
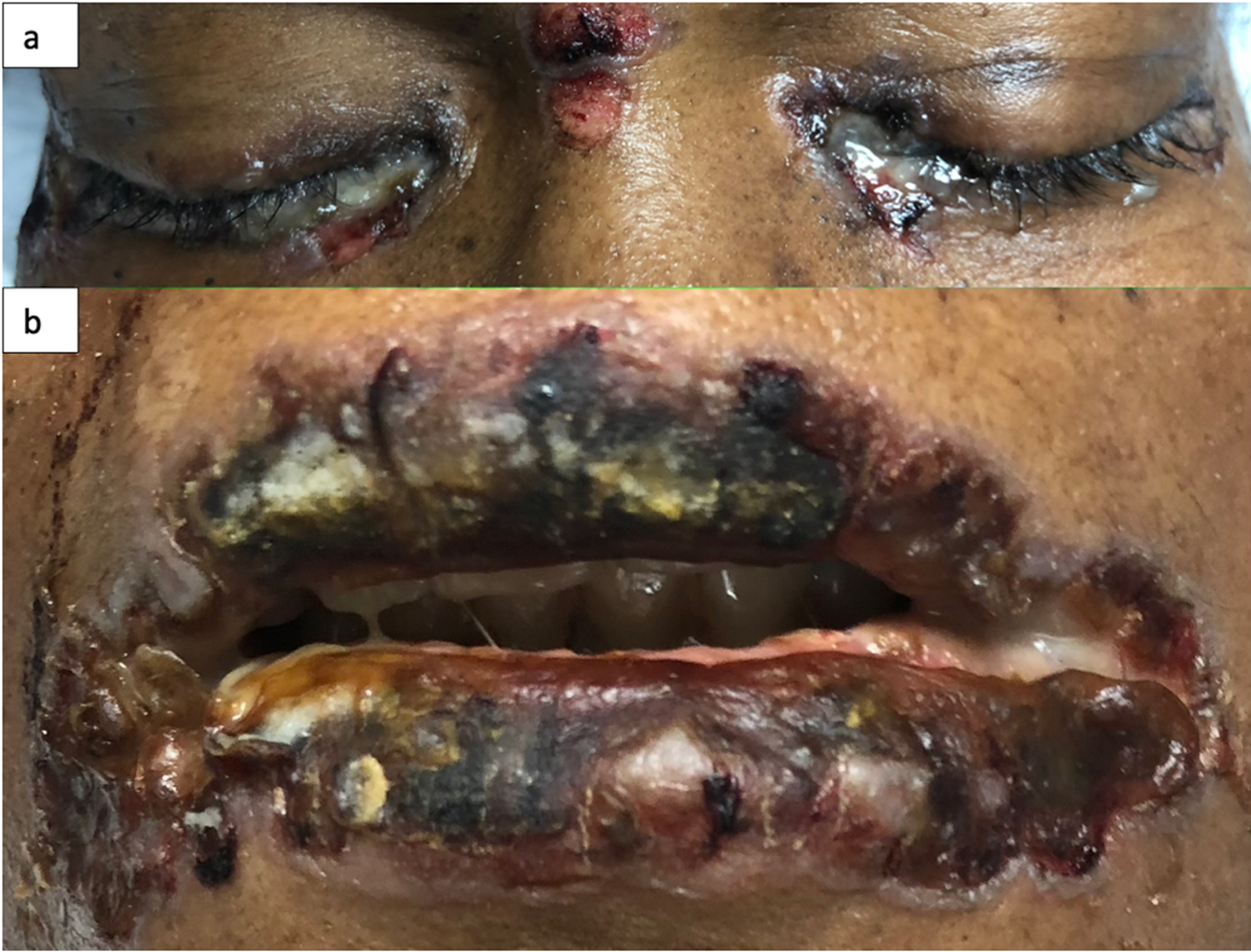
Figure 2: Oral and conjunctival mucosa with ulcerative desquamated hyperpigmented base and crusting over with sero-sanguineous fluid (a, b).
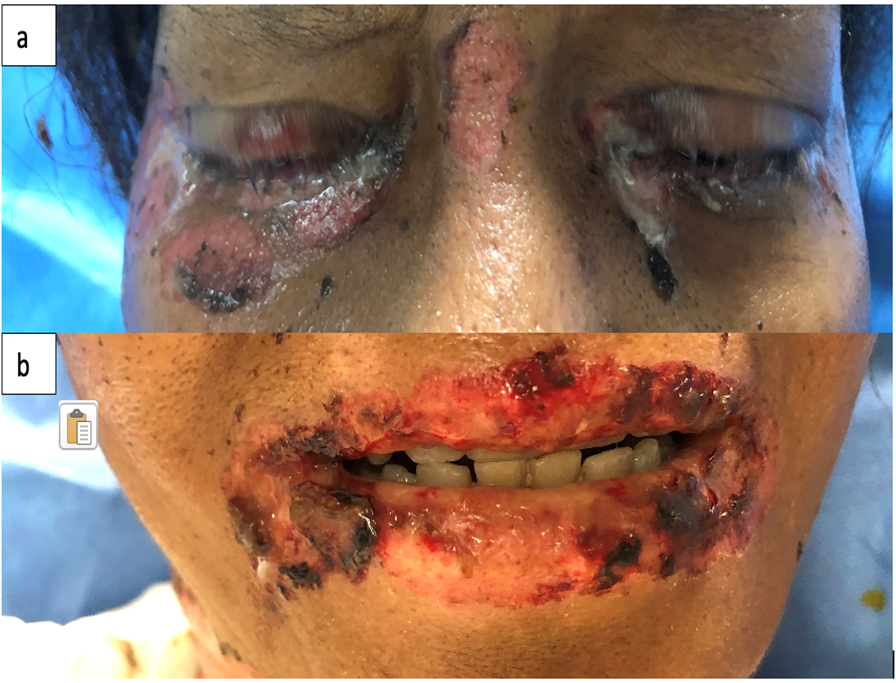
Figure 3: Skin with re-epithelialization and post inflammatory dyspigmentation (a, b).
Discussion
PV is a rare autoimmune disease and a challenging clinical diagnosis because it can be confused with a wide array of blistering diseases. Pemphigus vulgaris is subclassified as: Mucosa, Mucocutaneous and Cutaneous dominant. Mucosal manifestation most frequently involves the oral cavity, specifically the buccal (interior lining of cheeks) and palatine mucosa3. The oral cavity may be the only site of involvement for five months up to a year without any other skin manifestations7. There is an average of ten months in delay of diagnosis, usually after ulcer remains despite antibiotic, antifungal or antiviral topical medication6. Differential diagnosis of erosive oral lesions includes Infectious diseases (Herpes simplex, Impetigo), Drug-induced (Stevens-Johnson Syndrome, Toxic Epidermal Necrolysis), Vasculitis (Bechet disease) or Auto-immune diseases (Systemic Lupus Erythematosus or Oral lichen planus), among others8.
In white skin, PV’s skin lesions are described as scattered superficial erythematous flaccid, fragile blisters that evolve into scaly, crusted lesions, and erythematous ulcers when the oral or genital mucosa is involved. In darker skin tones, blisters may lack erythema and appear as hyperpigmented plaques with superficial erosion and hemorrhagic crusting, as seen in Figure 29. This change in appearance is mostly due to reactive hyperpigmentation occurring mostly in dark skin tone9. Reactive hyperpigmentation or post inflammatory hyperpigmentation is an acquired hyper-melanosis of leukotrienes (LT), such as LT-C4 and LT-D4, prostaglandins E2 and D2, thromboxane-2, interleukin (IL)-1, IL-6, tumor necrosis factor (TNF)-α, epidermal growth factor, and reactive oxygen species that develop melanocyte-stimulating properties. These melanocytes are transferred and placed unequally in the adjacent keratinocytes10. This clinical feature makes it difficult to recognize erythema and may mask a clinical diagnosis. In fact, this hyperpigmentation gives a purple shade to the lesion causing confusion and delay in diagnosis. Once disease has advance and coalesce to cover large body areas, plaques are characterized by pink base and peripheral hyperpigmentation as seen in Figure 19.
There are few epidemiological data concerning how skin color affects diagnosis, morbidity and mortality of PV. Saha et al (2013), found that ethnic groups with PV, such as Indo-Asian, are impacted by longer duration of disease when compared to White- Europeans, which included White British, other Europeans and Jewish patients11. In dermatological diseases, especially for those infectious, autoimmune and neoplastic disease, images are critical for the diagnoses. The lack of them, as it often happens in dark-skin tone pictures, leads to incorrect diagnoses and therefore, delay in medical treatment. Multiple studies have shown that people of color are underrepresented in both visual and textual content in preclinical medical materials and does not reflect the racial demographics of the U.S. population1. Dermatologists have complained about this issue. In fact, in a U.S. survey, 47% of all Dermatologist and Dermatology residents reported insufficient exposure and pertinent teaching materials concerning clinical presentations in dark skin12. This issue is not only manifesting as poorer patient’s satisfaction rate, but in poorer survival rates.
For example, the survival rate for melanoma, regardless of the stage at diagnosis, was lower among non-Hispanic black people than among non-Hispanic White people, mainly secondary to a delay in diagnosis13. While the factors that cause racial health-care disparities are complex, perhaps our deficient medical education regarding the scarce representation of dermatological diseases in dark-skinned tone patients and ethnics minorities is perpetuating this problem. Thus, defeating the purpose of preparing clinicians for treating a diverse population.
As for Pemphigus vulgaris, suspect it when a patient complains of a non-pruritic poor healing ulcer that started as a flaccid blister that may or may not be over an erythematous base. During the physical exam, it is imperative to verify and assess for palatine or buccal mucosa involvement. Be aware that black skin tone patients are more prone to Postinflammatory Hyperpigmentation (PIH), hence changing “the classic” appearance of PV.
This case report serves as an educational resource by providing images of a rare skin disease in people with dark skin with the purpose of filing major gaps in medical literature, highlighting the importance of timely recognizing PV in patients with dark skin, and creating awareness among physicians.
Acknowledgments
The authors have no conflict of interest to disclose. They declare that no funding sources are supporting this work.
Informed consent was obtained from the patient.
References
- Louie P, Wilkes R. Representations of race and skin tone in medical textbook imagery. Soc Sci Medicine. 2018; 202(1): 38-42. doi.o rg/10.1016/j.socscimed.2018.02.023.
- Kasperkiewicz M, Ellebrecht C, Takahashi H, et al. Pemphigus. Nat Rev Dis Primers. 2018; 3(17026): 1-40. doi:10.1038/nrdp.2017.26.
- Moncale-Campos A, Ezaguy-de Hollanda L, Makarem-Oliveira L, et al. Mucosal Manifestation of Pemphigus Vulgaris: A Key Sign for Early Diagnosis. Glob J Otolaryngol. 2019; 29(1): 4-8. DOI: 10.19080/GJO.2019.19.556002.
- Wertenteil S, Garg A, Strunk A, et al. Prevalence Estimates for Pemphigus in the United States. JAMA Dermatol. 2019; 155(5): 627-629. DOI: 10.1001/jamadermatol.2018.5954.
- Davenport S, Chen S and Miller A. Pemphigus Vulgaris: Clinicopathologic Review of 33 Cases in the oral Cavity. Int J PeriodonticsRestorative Dent. 2001; 21(1): 85-90. PMID: 11829040.
- Risser J, Lewis K and Weinstock M. Mortality of Bullous Skin Disorders From 1979 Through 2002 in the United States. Arch Dematol. 2009; 145(9): 1005-1008. DOI: doi:10.1001/archdermatol.2009.205
- Arpita R, Monica A and Venkatesh N. Oral Pemphigus Vulgaris: Case Report. Ethiop J Health Sci. 2015; (25)4: 367-372. DOI: http://dx.doi.org/10.4314/ejhs.v25i4.11
- Rashid H, Lamberts A, Diercks G, et al. Oral Lesions in Autoimmune Bullous Diseases: An Overview of Clinical Characteristics and Diagnostic Algorithm. Am J Clin Dermatol. 2019; 20(1): 847-861. DOI: doi.org/10.1007/s40257-019-00461-7.
- Tamazian S and Simpson C. Autoimmune Bullous Disease in Skin of Color: A Case Series. JAAD Case Reports. 2020; 6(1): 1173-1178. DOI: doi.org/10.1016/j.jdcr.2020.08.035.
- Davis E and Callender V. Postinflammatory Hyperpigmentation: A Review of the Epidemiology, Clinical Features, and Treatment Options in Skin of Color. J Clin Aesthet Dermatol. 2010; 3(7): 20-31. PMCID:PMC2921758.
- Saha M, Bhogal B, Black M, et al. Prognostic factors in Pemphigus Vulgaris and Pemphigus Foliaceus. BJD. 2014; 170(1): 116-122. Doi: 10.1111/bjd.12630.
- Buster K, Stevens E and Elmets C. Dermatologic Health Disparities. Dermatol Clin. 2012; 30(1): 54-64. Doi: 10.1016/j.det.2011.08.002.
- Culp M, Buchanan-Lunsford N. Melanoma Among Non-Hispanic Black Americans. Prev Chronic Dis. 2019; 16(6): e79. DOI: dx.doi.org/10.5888/pcd16.180640.